










Diagnóstico (2)
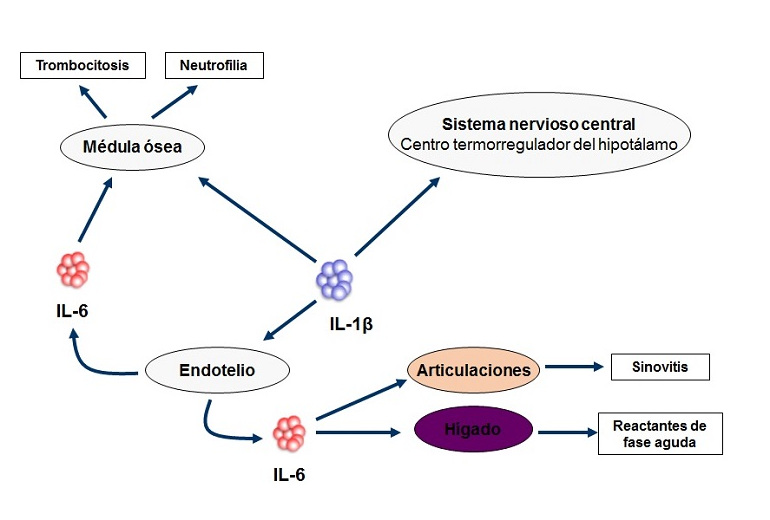
El síndrome de HIDS o síndrome de hiperinmunoglobulinemia IgD se trata de un síndrome autoinflamatorio hereditario producido por alteración en el gen MVK que codifica la enzima mevalonato quinasa. Esta enzima cataliza la conversión del ácido mevalónico en ácido fosfomevalónico. Como consecuencia de la reducción de su función, se genera una acumulación del metabolito inmediatamente anterior de la cadena (ácido mevalónico), que aparecerá en la orina durante los brotes. La disminución de metabolitos posteriores al bloqueo da lugar, por pérdida de una regulación negativa del inflamasoma, a una elevación de IL-1β responsable de la reacción inflamatoria.
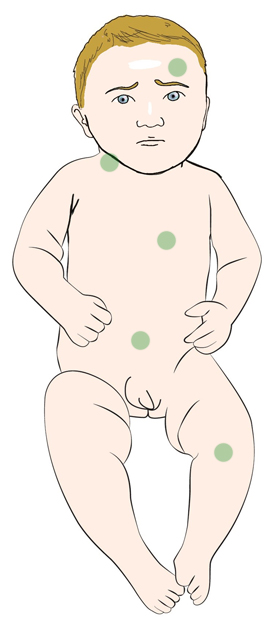
Se caracteriza por episodios frecuentes de fiebre que dura entre 3-7 días asociado a otras manifestaciones.
Suele diagnosticarse en la primera década de vida. Los episodios pueden desencadenarse con estrés, infecciones y, de forma característica, con inmunizaciones.
La gravedad de los episodios parece disminuir con el tiempo al reducirse los posibles desencadenantes.
Para llegar a su diagnóstico, puede realizarse un estudio genético o determinar ácido mevalónico en orina. Los niveles de IgD suelen estar elevados, lo que dio originalmente el nombre al cuadro.
El tratamiento es sintomático, pudiéndose emplear antiinflamatorios no esteroideos o corticoides durante los episodios agudos y, en aquellos pacientes con brotes intensos o muy frecuentes, fármacos que bloqueen la IL-1 (anakinra, canakinumab).

 No hay comentarios
No hay comentarios 