










Enfermedad de Wilson (2)
El diagnostico final fue de EW en fase presintomática.
La EW es consecuencia de un trastorno del metabolismo del cobre que favorece su depósito en hígado, cerebro, riñón y otros órganos, con un efecto tóxico variable y daño celular consecuente. Su base es genética, por mutaciones en el gen ATP7B, y su herencia autosómica recesiva. Su prevalencia aproximada es 1 por cada 30-50 000 habitantes. La incidencia de portadores heterocigotos es de 1 por cada 90 individuos.
La utilidad de los análisis de mutaciones del gen ATP7B, del cual hay descritas más de 500, es el diagnóstico diferencial entre pacientes heterocigotos frente a pacientes presintomático, sobre todo en edades tempranas de la vida. La mutación más frecuente en Europa Central y del Este es His106Glu (H1069Q) presente en el 50-80% de los pacientes y en España la mutación Met645Arg se demuestra en casi la mitad de los enfermos. No se ha demostrado correlación entre genotipo y fenotipo.
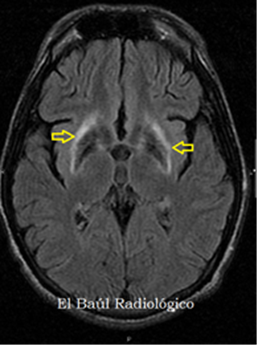
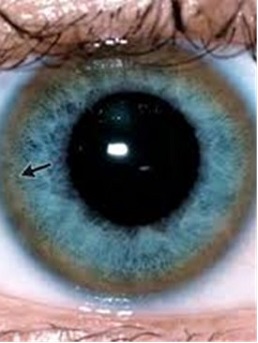
Su depósito hepático provoca daño celular en el hepatocito que se manifiesta desde afectación hepática aislada con disfunción asintomática (habitual en niños) hasta cirrosis o insuficiencia hepática aguda grave. La afectación neurológica (más frecuente en niños mayores y adultos) por depósito de cobre en los ganglios basales cursa con síntomas extrapiramidales o cerebelosos en forma de disartria, temblor, manifestaciones psiquiátricas… y suele acompañarse de depósito de cobre en la membrana corneal de Descemet (anillo de Kayser-Fleischer). Otras alteraciones incluyen: afectación renal en forma de tubulopatía y nefrolitiasis, alteraciones esqueléticas (osteoporosis…), anemia hemolítica, Coombs negativa por depósito de cobre en los hematíes, pancreatitis y miocardiopatía.
 No hay comentarios
No hay comentarios 