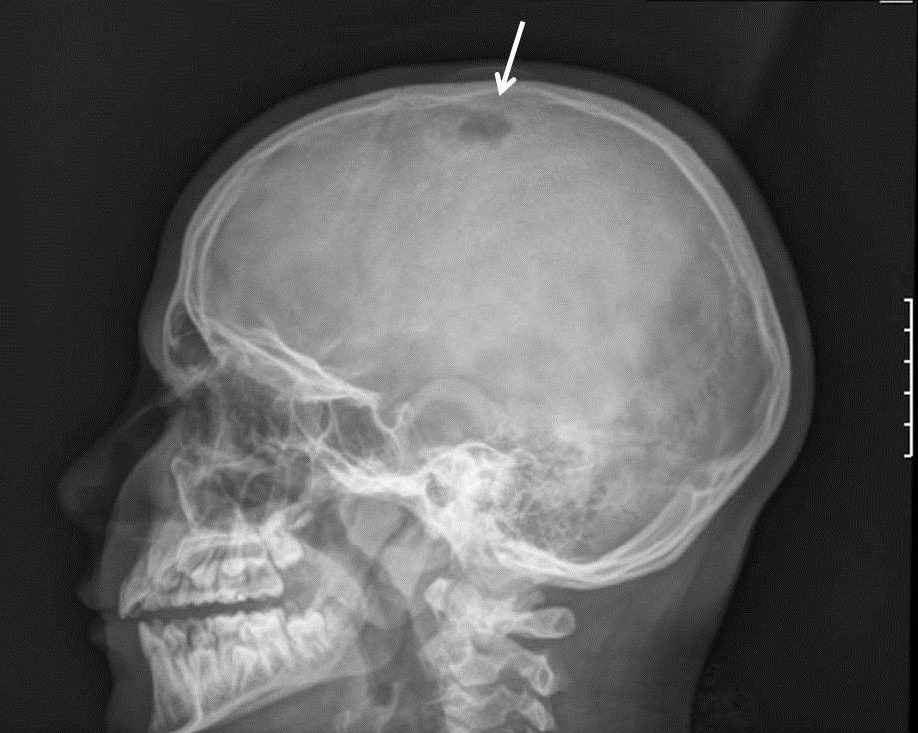
Respuesta correcta. Por su localización, la edad de la paciente y las características morfológicas de la lesión, la primera posibilidad diagnóstica que nos planteamos es un granuloma eosinófilo. El granuloma eosinófilo (GE) es una enfermedad infrecuente que suele afectar de forma predominante a varones y aparece con mayor frecuencia durante la infancia y la adolescencia1,2.
Bibliografía
- Gómez-Ruiz C, Buxadé-Martí I, Hinojosa-Bareas G. Granuloma eosinófilo óseo. SEMERGEN. 2011;37:573-5.
- Abad Royo JM, Pérez Sánchez A, Chamizo Garcia JJ, Jiménez F, Martínez P. Granuloma eosinófilo en el hueso temporal. ORL Aragón. 2002;5:11-5.
Es la más benigna de las tres variantes clínicas de histiocitosis X (granuloma eosinófilo, enfermedad de Hand-Schüller-Christian y enfermedad de Letterer-Siwe)2. El granuloma eosinófilo presenta afectación ósea, pudiendo ser monostótico o poliostótico1, sin presentar afectación sistémica, a diferencia de las otras dos entidades de la histiocitosis X. Las localizaciones más frecuentes del granuloma eosinófilo son el cráneo, huesos largos, costillas, vértebras y la mandíbula1. Se trata de una lesión lítica en el hueso desarrollada por una anormal proliferación de los histiocitos, que puede erosionar la cortical de la bóveda craneal y presentar un crecimiento intracraneal que puede manifestarse con focalidad neurológica. Se presenta como una lesión exofítica, de límites bien definidos, de consistencia dura, sin crepitación ni alteración de la piel adyacente. En la radiografía de cráneo se presenta como una lesión radiolúcida de características líticas, redondeada u ovalada, y con bordes esclerosados1, tal y como observamos en nuestro caso. Para completar el estudio de la extensión intracraneal, realizamos una tomografía axial computarizada craneal donde se objetivó una lesión lítica ovalada de 1 × 1,5 × 1 cm de contornos irregulares pero bien definidos, con defecto en ambas corticales con importante componente de masa de partes blandas extracraneales y un componente intracraneal pequeño. Posteriormente se realiza biopsia, compatible con histiocitosis de células de Langerhans (GE) con expresión de CD1a, tratándose con corticoide depot local. En revisiones posteriores se objetiva la desaparición completa de la tumoración, sin volver a presentar nuevas crisis de ausencia.











 Más información
Más información
 No hay comentarios
No hay comentarios 